3.10.4.4 Avogadro2 — программа просмотра и редактирования молекул
 Скачать документ
Скачать документ
Установка
Описание программы
Пример работы
Окружение
- Версия ОС: 8
- Конфигурация ОС: Рабочая станция
- Редакция ОС: Стандартная, Образовательная
- Архитектура: x86_64
- Версия ПО: avogadro2-ru-1.97.0-1
Avogadro2 — редактор и визуализатор молекул, предназначенный для кроссплатформенного использования в вычислительной химии, молекулярном моделировании, биоинформатике, материаловедении и смежных областях. Функционал программы можно расширить с помощью плагинов.
С помощью Avogadro2 можно легко построить или изменить структуру молекулы, при этом программа обеспечивает отличное качество отображения молекул и белковых структур. Приложением поддерживается просмотр и редактирование трёхмерных структур большинства наиболее популярных в вычислительной химии форматов (таких как *.cml, *.cif, *.mol, *.sd, *.sdf, *.pdb, *.ent, *.vis, *.xyz и пр).
Вы можете подробнее ознакомиться с работой программы avogadro, просмотрев наши обучающие видео:
-
на RuTube — Avogadro. Программа 3D моделирования молекул;
-
в Яндекс.Дзен — Avogadro. Программа 3D моделирования молекул;
-
в VK Видео — Avogadro. Программа 3D моделирования молекул.
На наших каналах вы также сможете найти много другой полезной информации.
Установка
Установить программу можно либо через графический менеджер пакетов, либо через терминал.
Для установки программы через графический менеджер пакетов dnfdragora перейдите в «Главное меню» — «Администрирование» — «Управление пакетами dnfdragora», выполните поиск необходимого пакета по ключевому слову «avogadro2-ru» и отметьте флагом пакет последней версии. После этого нажмите кнопку «Применить» и дождитесь окончания установки пакетов.
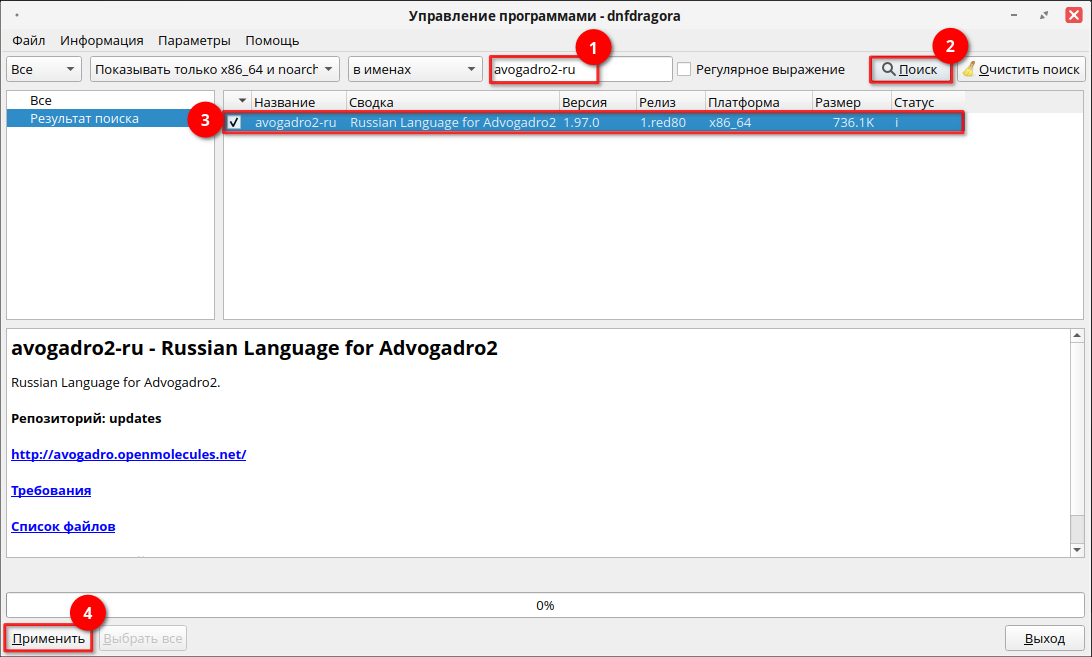
Для установки программы через терминал выполните следующую команду (потребуются права администратора):
sudo dnf install avogadro2-ru
Дальнейшая работа в программе выполняется от локального пользователя.
Для запуска программы перейдите в «Главное меню» — «Образовательные» — «Avogadro2» или выполните команду в терминале:
avogadro2
Описание программы
Интерфейс программы стандартный и состоит из нескольких типовых частей.
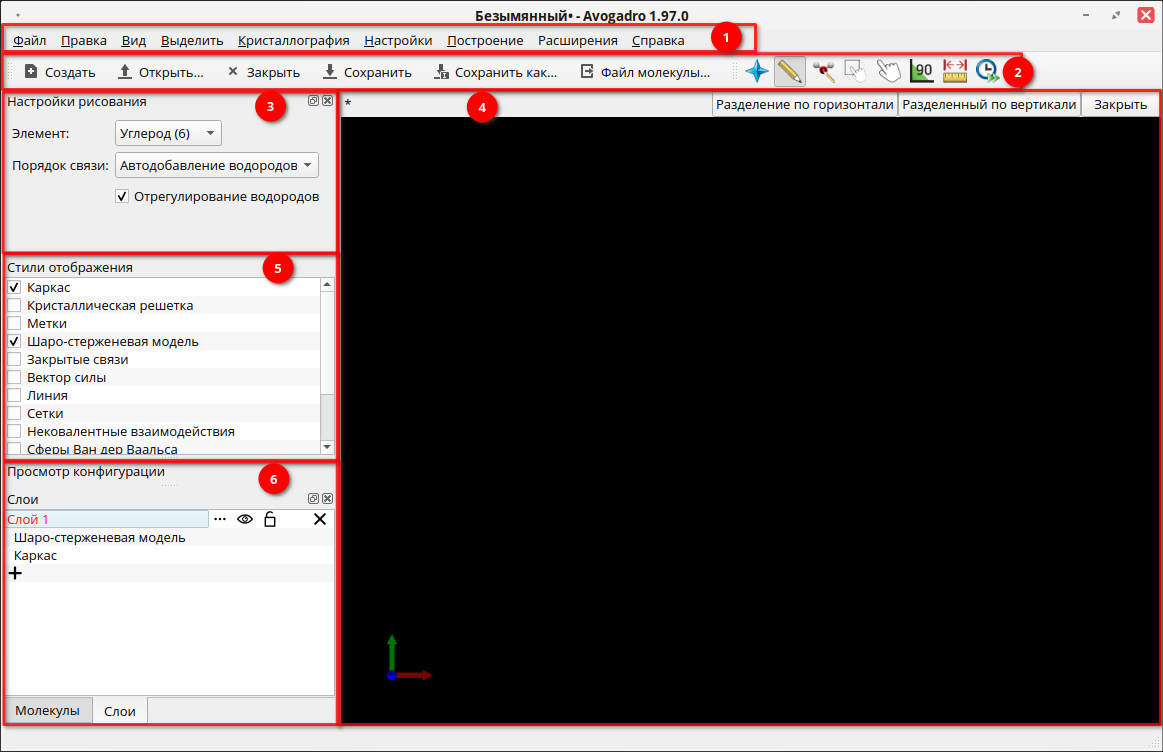
1. Основное меню программы. Позволяет выполнять различные действия с создаваемым документом, а также настраивать как саму программу, так и панель инструментов.
2. Панель инструментов. Это стандартная панель инструментов с кнопками быстрого доступа к основным функциям программы. Эти панели можно скрывать и отображать через основное меню. По умолчанию отображается Главная панель и Инструменты.
В меню настройки доступно много панелей, но они нужны при более глубоком изучении приложения.
3. Панель рисования. Здесь отображаются опции и действия, которые относятся к выбранному в данный момент инструменту. Инструменты можно выбирать через панель инструментов.
4. Основное рабочее поле. В данном поле происходит весь процесс рисования и анимации моделей молекул.
5. Панель стилей отображения модели. Эта панель позволяет менять способ отображения молекулы.
6. Панель просмотра конфигурации. Здесь отображаются опции и действия, которые относятся к выбранному в данный момент инструменту. Инструменты можно выбирать через панель инструментов.
Пример работы
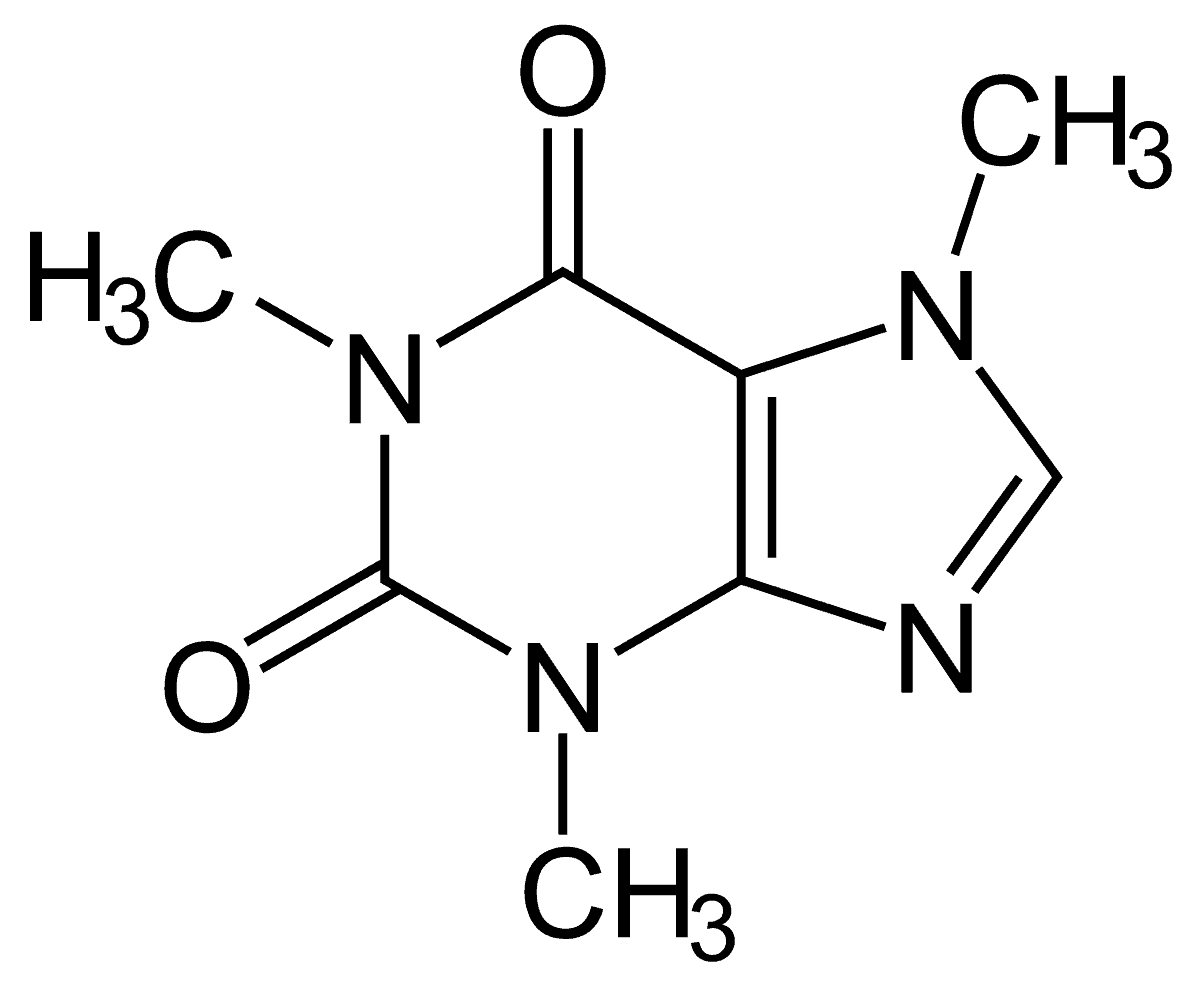
Рассмотрим создание молекулы на примере молекулы кофеина, которая имеет формулу C8H10N4O2 и имеет структуру следующего вида:
Сначала выберите атом кислорода и оставьте одинарную связь. Связи в процессе можно добавлять.
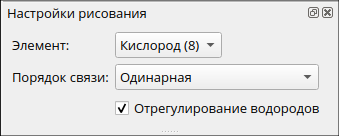
Разместите атом на рабочем поле. В соответствии с настройками по умолчанию выстраивается молекула воды (H2O).
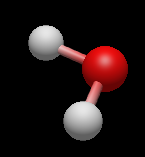
Далее выберите атом углерода. На рабочем поле наведите курсор на атом кислорода и, удерживая ЛКМ, переместите курсор в сторону, чтобы создать химическую связь. Затем отпустите кнопку мыши для завершения соединения.
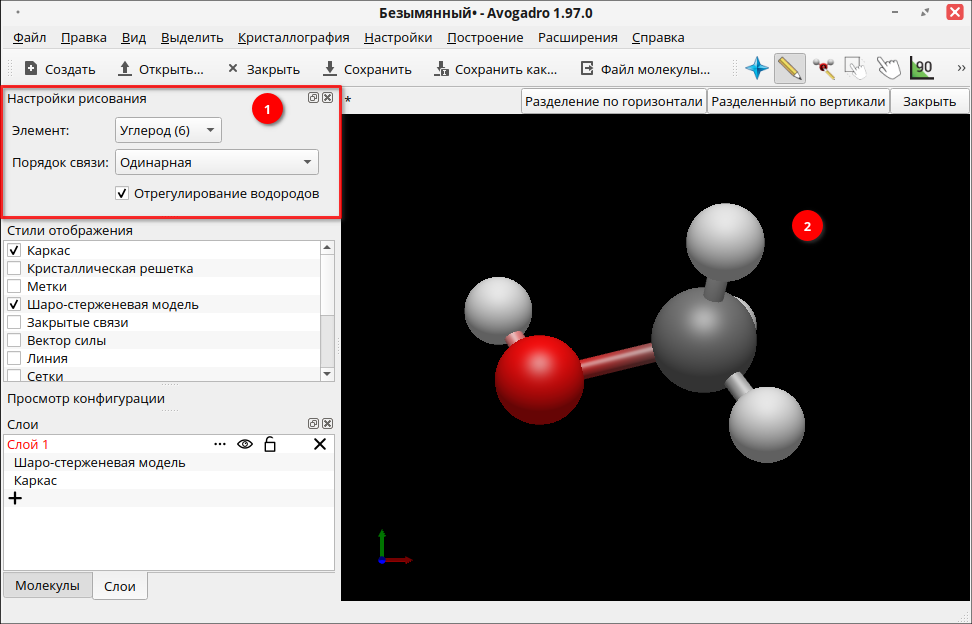
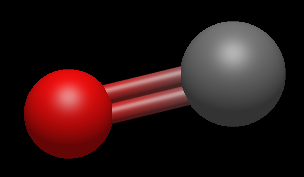
Наведите курсор на связь между кислородом и углеродом и добавьте еще одну молекулярную связь:
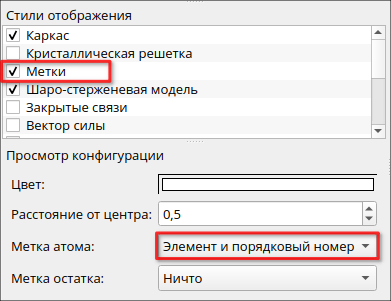
Стили отображения можно настроить на панели Просмотр конфигурации. Например, в пункте Метки атома можно добавить в созданную молекулу подписи использующихся химических элементов, выбрав «Элемент и порядковый номер».
Добавьте по очереди остальные атомы. В результате получается молекула такого вида:
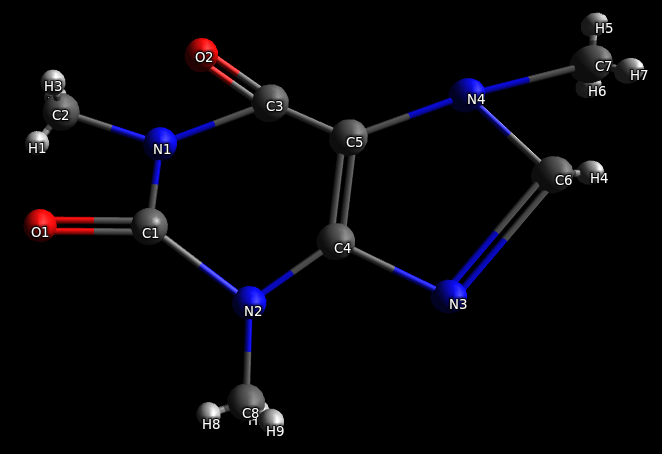
Необходимо оптимизировать длины и углы связей. Для этого в основном меню выбираем соответствующий пункт «Расширения» — «Открыть Babel» — «Оптимизировать геометрию».
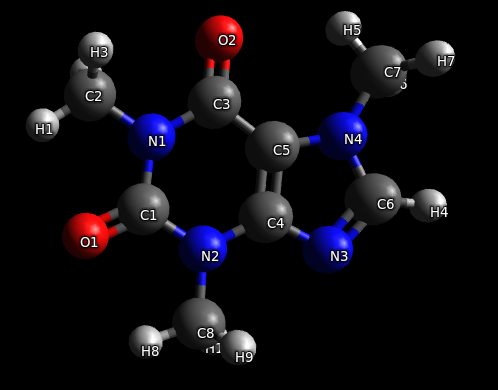
После молекула выглядит правильно, поскольку расстояние между атомами и углы между связями установлены в соответствии со справочными данными.
В Настройках перемещения можно рассмотреть молекулу со всех сторон.
Используя Стили отображения, также существует возможность, например, отобразить сферы Ван-дер-Ваальса.
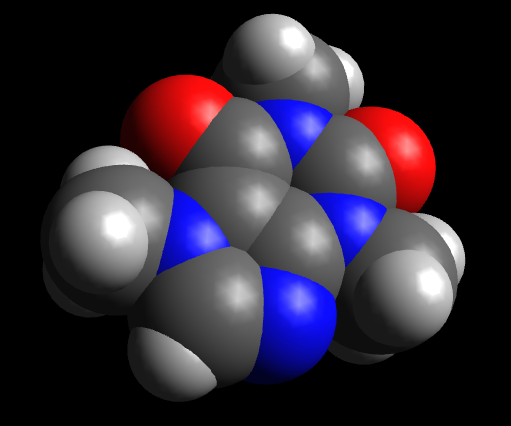
На этом создание молекулы завершено.
Дата последнего изменения: 29.08.2025
Если вы нашли ошибку, пожалуйста, выделите текст и нажмите Ctrl+Enter.