3.10.4.7 Gabedit — графический интерфейс для пакетов вычислительной химии
 Скачать документ
Скачать документ
Установка
Запуск Gabedit
Первоначальная настройка
Описание программы
Пример работы
Окружение
- Версия ОС: 8
- Конфигурация ОС: Рабочая станция
- Редакция ОС: Стандартная, Образовательная
- Архитектура: x86_64
- Версия ПО: gabedit-2.5.1
Gabedit — это графический интерфейс (GUI) для различных программ в области вычислительной химии. Он поддерживает такие программы, как Gaussian, GAMESS, VASP и другие. Gabedit представляет средство для визуализации молекулярных структур, подготовки входных данных и обработки выходных данных.
Основные характеристики:
-
Gabedit может создавать входные файлы для GAMESS(US), GAUSSIAN, MOLCAS, MOLPRO, MPQC, MOPAC, Orca, PCGamess и Q-Chem;
-
в комплект входит «Конструктор молекул» для быстрого создания эскизов молекул и их изучения в трёхмерном пространстве;
-
Gabedit может графически отображать результаты расчётов:
-
молекулярные орбитали;
-
поверхности с плотностью электронов, электростатическим потенциалом, плотностью экранирования ЯМР, ИК- и КР-спектры и другими свойствами;
-
поверхности могут отображаться в сплошном, полупрозрачном и сетчатом режимах. Они могут быть раскрашены с помощью отдельного свойства;
-
контуры (с цветовой кодировкой), плоскости с цветовой кодировкой, диполь. Оси XYZ и главные оси молекулы;
-
анимация нормальных режимов, соответствующих частотам колебаний;
-
анимация вращения геометрии, поверхностей, контуров, плоскостей с цветовой кодировкой, XYZ и главных осей молекулы;
-
анимация контуров, анимация плоскостей с цветовым кодированием;
-
-
Gabedit может создавать файлы povray для геометрии, поверхностей, контуров, плоскостей с цветовой кодировкой;
-
доступен экспорт в различные форматы файлов, такие как BMP, JPEG, PNG, PPM и PS;
-
Gabedit может автоматически генерировать серию изображений для анимации (вибрация, схождение геометрии, вращение, контуры, плоскости с цветовой кодировкой);
-
моделирование отжига с помощью молекулярной динамики;
-
реализовано несколько инструментов кристаллографии.
Установка
Установить программу можно либо через графический менеджер пакетов, либо через терминал.
Для установки программы через графический менеджер пакетов dnfdragora перейдите в «Главное меню» — «Администрирование» — «Управление пакетами dnfdragora», выполните поиск необходимого пакета по ключевому слову «gabedit» и отметьте флагом пакет последней версии. После этого нажмите кнопку «Применить» и дождитесь окончания установки пакетов.
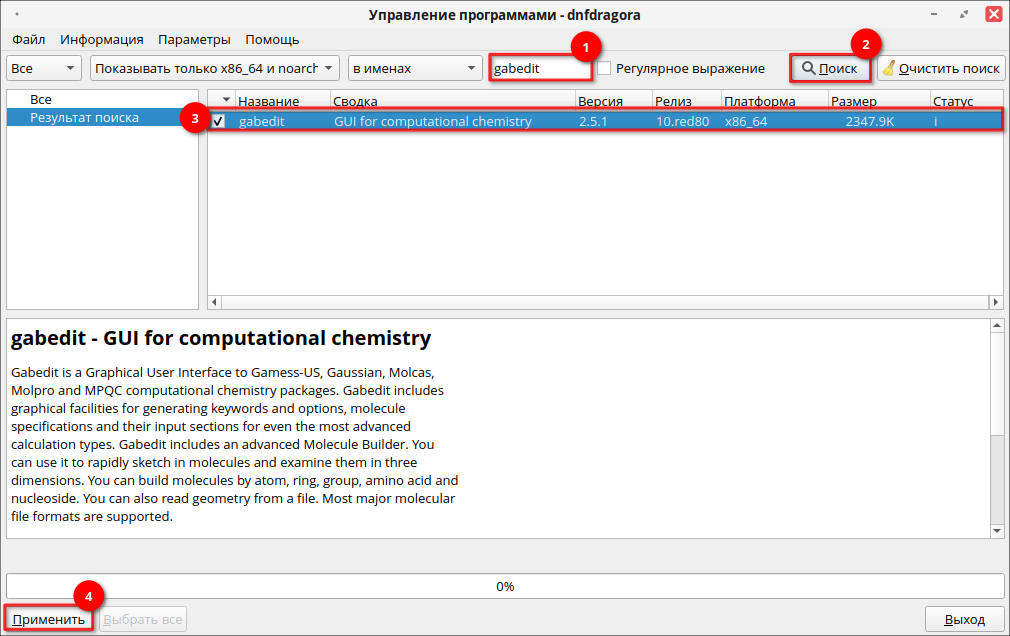
Для установки программы через терминал выполните следующую команду (потребуются права администратора):
sudo dnf install gabedit
Дальнейшая работа в программе выполняется от локального пользователя.
Запуск Gabedit
После установки программу можно запустить из «Главного меню» — «Образовательные» — «Вычислительная химия Gabedit» или с помощью команды в терминале:
gabedit
Первоначальная настройка
При первом запуске Gabedit автоматически откроется мастер начальной настройки. Для перехода к следующему шагу нажмите кнопку «Continue».

Для корректной работы приложения необходимо создается конфигурационный каталог /home/user/.gabedit-2.5.1. Он будет содержать все пользовательские параметры и служебные файлы. Для получения более подробной информации о выбранном элементе нажмите на один из файлов.

В процессе начальной настройки выполняется создание вспомогательных каталогов.

Настройка параметров отображения атомов.

Выбор сетевых протоколов, необходимых для взаимодействия с внешними расчетными программами (можно изменить позже в разделе «Settings» — «Preferences»).
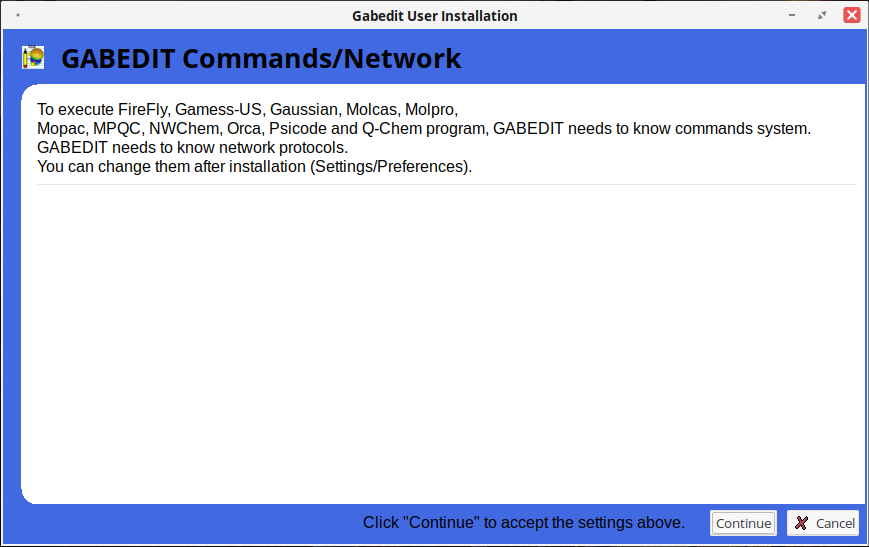
Настройка параметров визуализации: шрифты, цвет текста, фон.
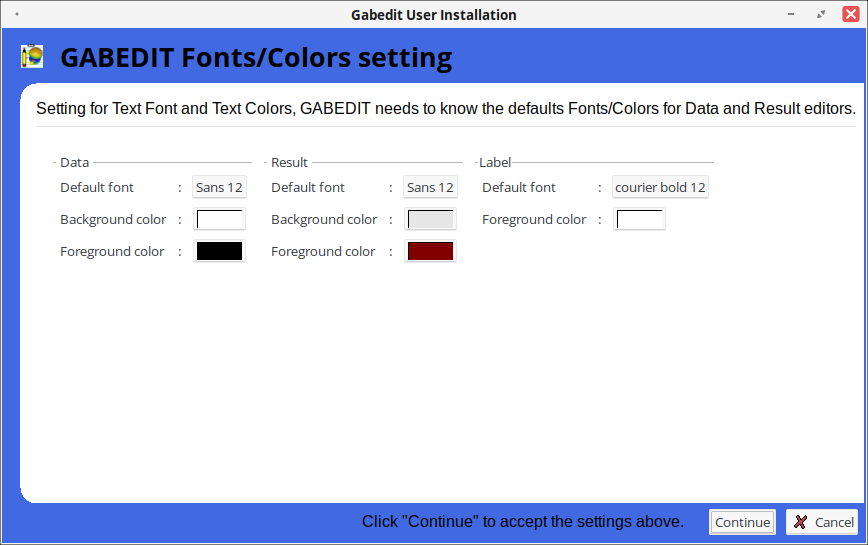
Настройка стилей отображения поверхностей Gabedit (плотность, орбитали).
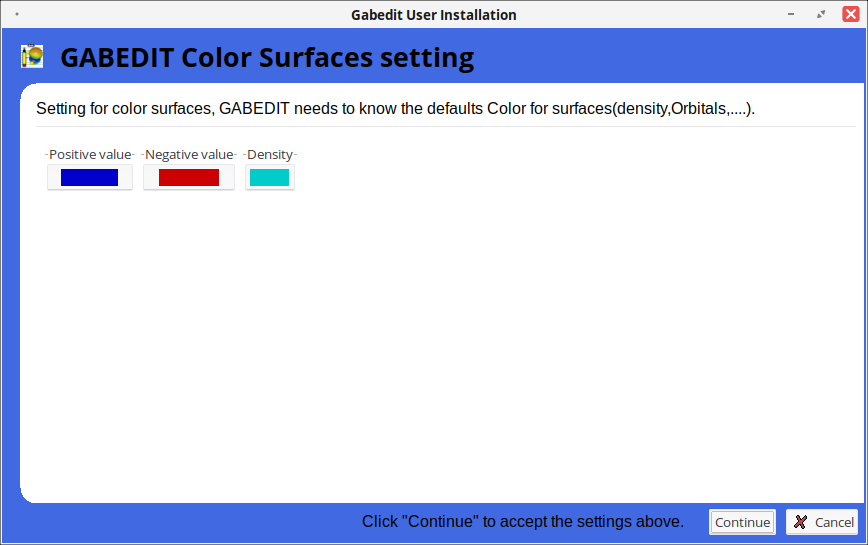
Создание базовых файлов конфигурации molpro.
Создание базовых файлов конфигурации molcas.
Создание базовых файлов конфигурации mpqc.
После завершения всех этапов конфигурации нажмите «Continue» для запуска Gabedit с установленными параметрами.

Программа будет загружена с сохранёнными настройками и станет доступна для дальнейшей работы.

Описание программы
Главное окно программы выглядит следующим образом:
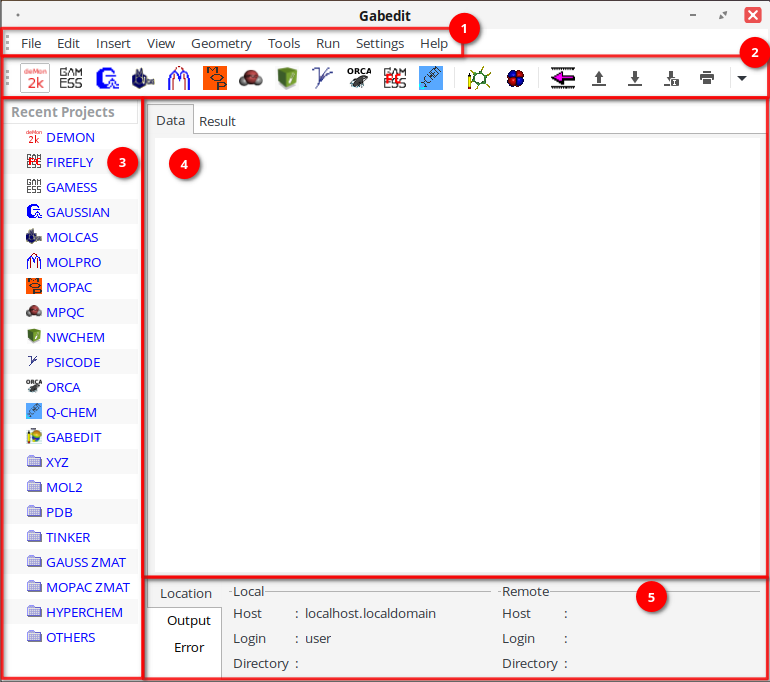
1. Основное меню расположено в верхней части окна и включает следующие разделы:
-
File — работа с файлами (создание, открытие, сохранение проектов);
-
Edit — базовые инструменты редактирования молекул;
-
Insert — вставка шаблонов или фрагментов для расчетных пакетов (Gaussian, Molcas, Molpro);
-
View — настройка отображения молекул, видов и визуализации;
-
Geometry — инструменты редактирования геометрии (ручное редактирование, автогенерация, взаимодействие с расчетными пакетами);
-
Tools — вспомогательные инструменты: запуск процессов, пакетная обработка, интеграция с Open Babel;
-
Run — запуск расчетов и работа с выходными файлами расчетных программ;
-
Settings — настройка параметров программы;
- Help — справочная информация о программе.
2. Панель инструментов содержит кнопки быстрого доступа к часто используемым функциям (вращение и перемещение молекулы, выбор и редактирование атомов, удаление и добавление элементов, запуск расчетов и визуализация результатов).
![]() — новый входной файл DeMon;
— новый входной файл DeMon;
![]() — новый входной файл Gamess;
— новый входной файл Gamess;
![]() — новый входной файл Gaussian;
— новый входной файл Gaussian;
![]() — новый входной файл Molcas;
— новый входной файл Molcas;
![]() — новый входной файл Molpo;
— новый входной файл Molpo;
![]() — новый входной файл Mopac;
— новый входной файл Mopac;
![]() — новый входной файл MPQC;
— новый входной файл MPQC;
![]() — новый входной файл NWChem;
— новый входной файл NWChem;
![]() — новый входной файл psicode;
— новый входной файл psicode;
![]() — новый входной файл Orca;
— новый входной файл Orca;
![]() — новый входной файл FireFly;
— новый входной файл FireFly;
![]() — новый входной файл Q-Chem;
— новый входной файл Q-Chem;
![]() — нарисовать геометрию;
— нарисовать геометрию;
![]() — отображение геометрии/орбиталей/плотности;
— отображение геометрии/орбиталей/плотности;
![]() — включить файл;
— включить файл;
![]() — открыть файл;
— открыть файл;
![]() — сохранить файл;
— сохранить файл;
![]() — сохранить как;
— сохранить как;
![]() — печать;
— печать;
![]() — вырезать;
— вырезать;
![]() — копировать;
— копировать;
![]() — вставить;
— вставить;
![]() — найти;
— найти;
![]() — выбрать все;
— выбрать все;
![]() — запустить программу;
— запустить программу;
![]() — преобразовать единицы измерения;
— преобразовать единицы измерения;
![]() — выйти из программы;
— выйти из программы;
![]() — справка о программе.
— справка о программе.
3. Панель навигации расположена слева, отображает загруженные молекулы, открытые проекты или связанные файлы. Позволяет быстро переключаться между объектами в текущей сессии.
4. Рабочая область — центральная часть окна, в которой выполняется основная работа: редактирование структуры молекулы, добавление/удаление атомов и связей, изменение координат, зарядов и других свойств атомов, визуализация молекулярной геометрии.
5. Панель состояния — находится в нижней части интерфейса, отображает вспомогательные уведомления и сообщения об ошибках.
GAMESS-US (General Atomic and Molecular Electronic Structure System) — утилита для вычислительной квантовой химии, которая позволяет проводить расчёты с использованием различных методов. Результаты расчётов сохраняются в текстовом файле специального формата. Координаты молекул, полученные в GAMESS-US, можно легко преобразовать в другие форматы с помощью утилиты Open Babel.
Gaussian — это комплекс утилит для расчёта структуры и свойств молекулярных систем как в газовой, так и в конденсированной фазах. Пакет включает широкий спектр методов вычислительной и квантовой химии, а также молекулярного моделирования. Gaussian позволяет генерировать cube-файлы, которые описывают объемные данные и расположение атомов в молекулярной системе.
Molcas — это коммерческое приложение для вычислительной химии, разработанное в рамках международного сотрудничества. Основное внимание в Molcas уделяется методам расчёта электронной структуры молекул, а также использованию библиотеки для параллельных вычислений Global Arrays.
Molpro — это набор утилит, основанных на библиотеке параллельных вычислений Global Arrays, предназначенный для высокоточных расчётов в квантовой химии. При разработке Molpro акцент делается на точность вычислений и детальное рассмотрение корреляции электронов через взаимодействие конфигураций многовариантности.
MPQC (Massively Parallel Quantum Chemistry) — это пакет утилит, предназначенный для параллельных вычислений в области квантовой химии и молекулярного моделирования. MPQC является открытым аналогом таких пакетов, как GAMESS и Gaussian.
OpenMopac — это утилита, используемая в вычислительной химии для реализации полуэмпирических методов квантовой химии. Она представляет собой свободную версию MOPAC.
Orca — это пакет утилит для вычислительной квантовой химии, ориентированный на расчёт больших молекул и комплексов с переходными металлами, а также на UV-Vis и ИК-спектроскопию. Результаты вычислений сохраняются в специализированном формате, но могут быть преобразованы в cube-файлы и формат XYZ, поддерживаемый большинством приложений для трёхмерного молекулярного моделирования.
Firefly — это набор утилит для квантово-химических расчётов, основанный на GAMESS-US.
Q-Chem — это пакет, предлагающий передовые инструменты неэмпирической химии для точного прогнозирования молекулярных структур, реактивности, колебательных и электронных спектров, а также спектров ядерного магнитного резонанса. Q-Chem включает интегрированный графический интерфейс с возможностями визуализации и генерации ввода, а также обширный набор функций и методов корреляции.
Gabedit поддерживает наиболее популярные форматы молекул и белковых структур (таких как *.cml, *.cif, *.mol, *.sd, *.sdf, *.pdb, *.ent, *.vis, *.xyz и пр). Gabedit также поддерживает импорт различных графических форматов (таких как PNG, JPEG, BMP, PPM и др.) и может автоматически генерировать серию изображений для анимации и многое другое.
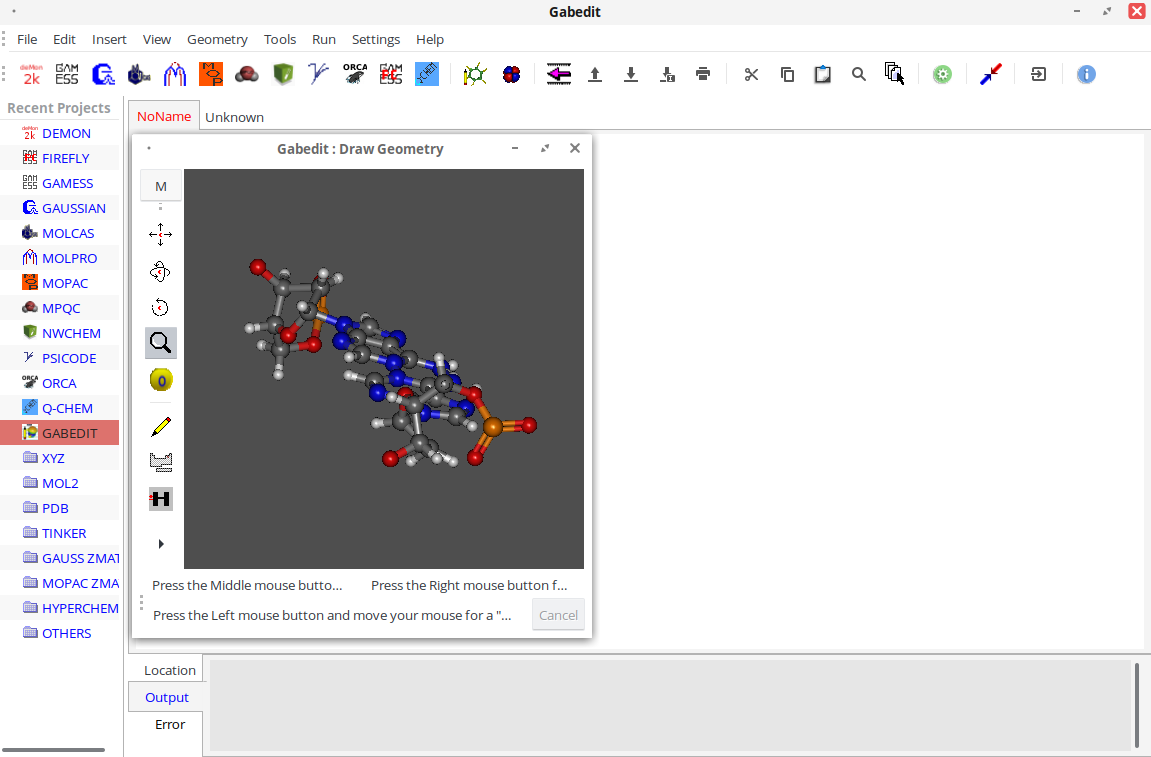
Пример работы
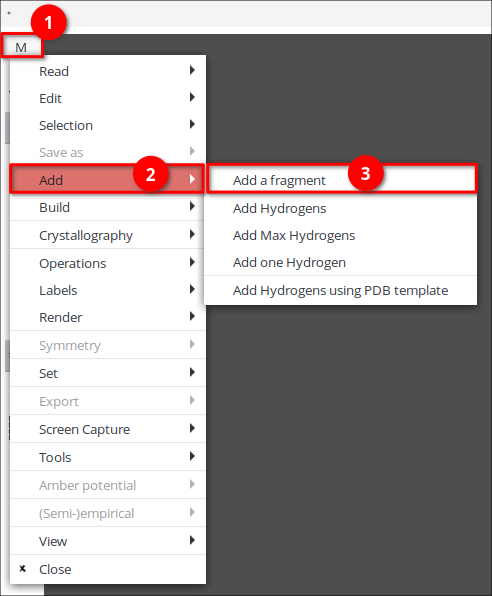
1. Для построения молекулы на панели инструментов выберите инструмент ![]() .
.

2. В открывшемся окне выберите в меню (1) — «Add» (2) — «Add a fragment» (3).
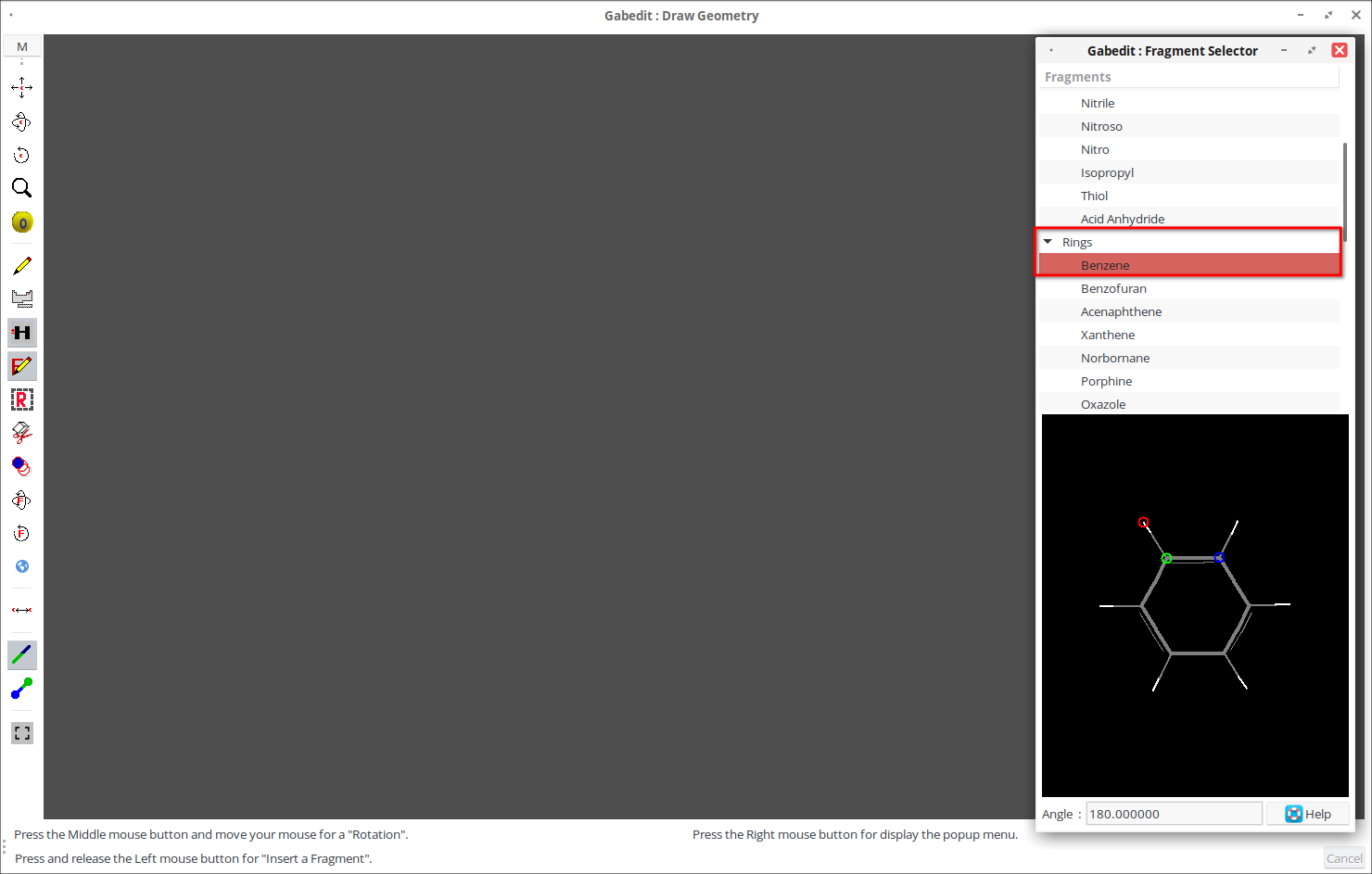
3. В списке фрагментов выберите необходимый элемент, например, «Rings» — «Benzene».
4. Перенесите фрагмент в область рисования.
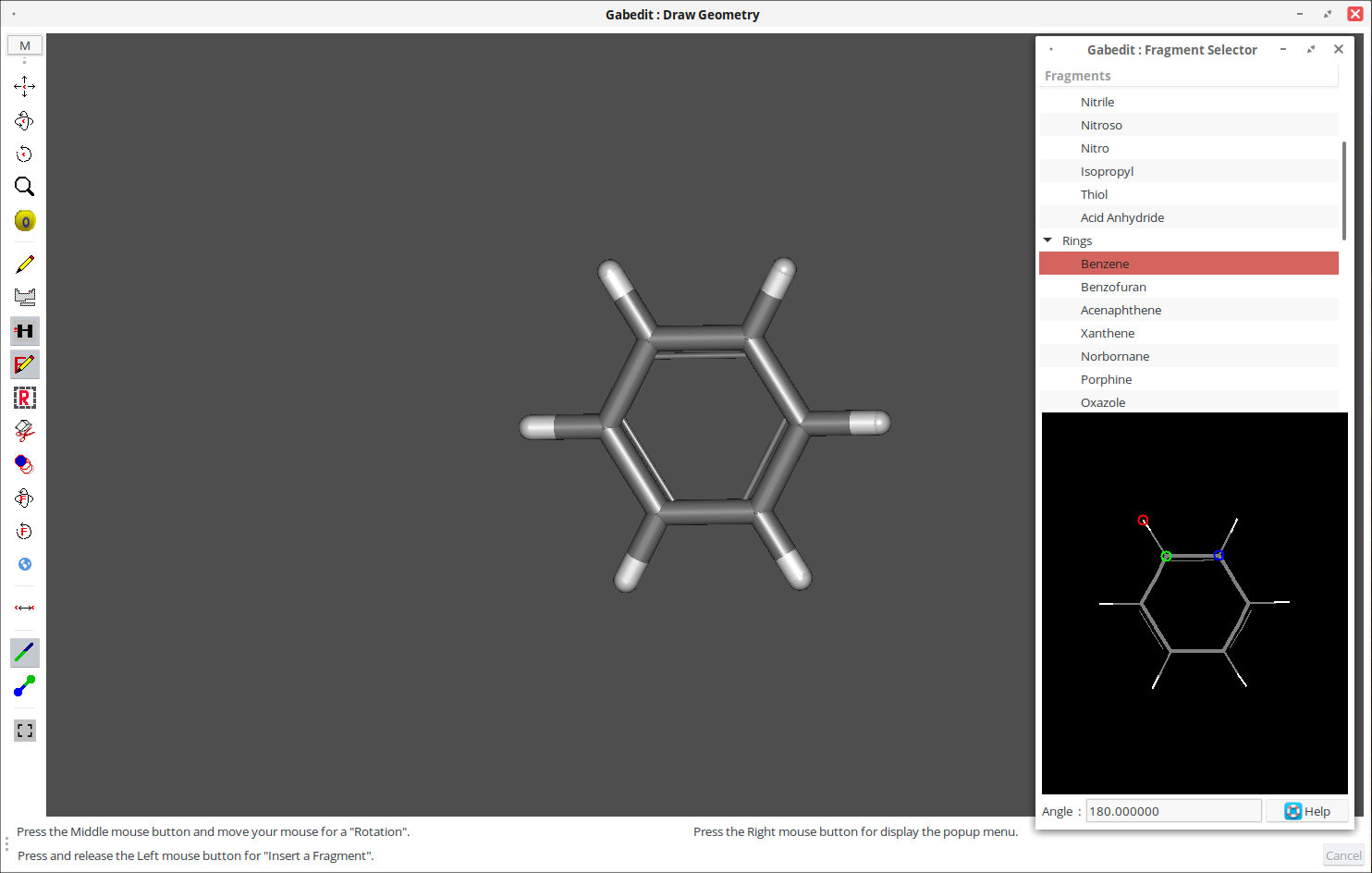
5. При необходимости добавьте дополнительные фрагменты аналогичным способом.
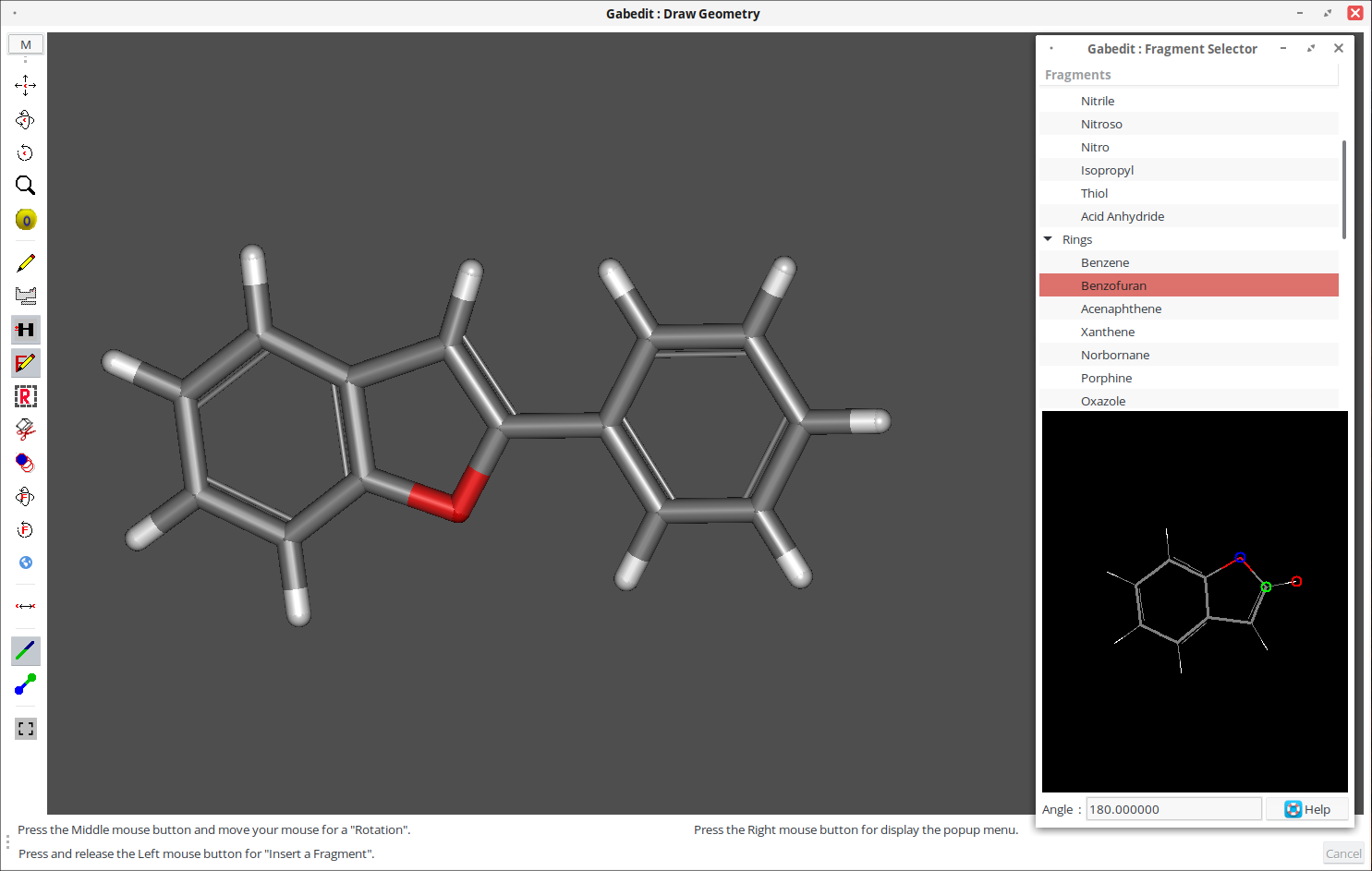
6. В меню выберите пункт «Save as» — «Gabedit file».
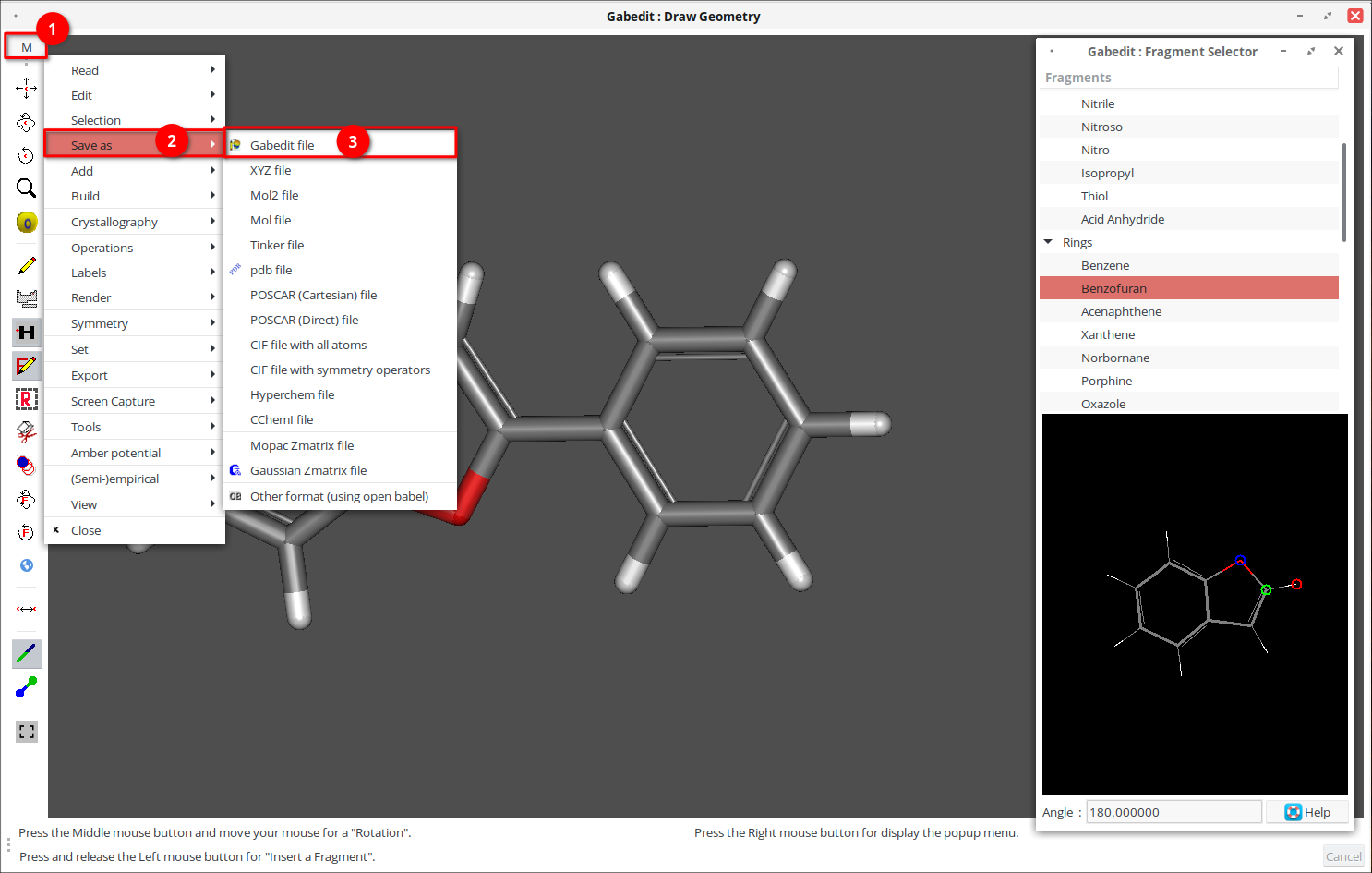
7. Укажите имя файла и сохраните его с расширением *.gab, затем закройте окно рисования геометрии.
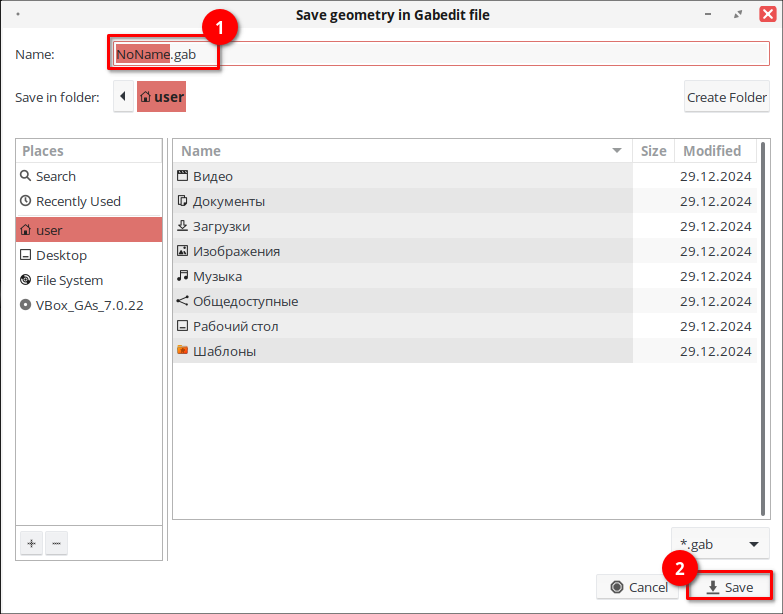
8. В навигационной панели выберите «ORCA», затем на панели инструментов нажмите «Open a file». Установите отображение всех файлов (*), выберите сохранённый файл с расширением *.gab и нажмите «Open».
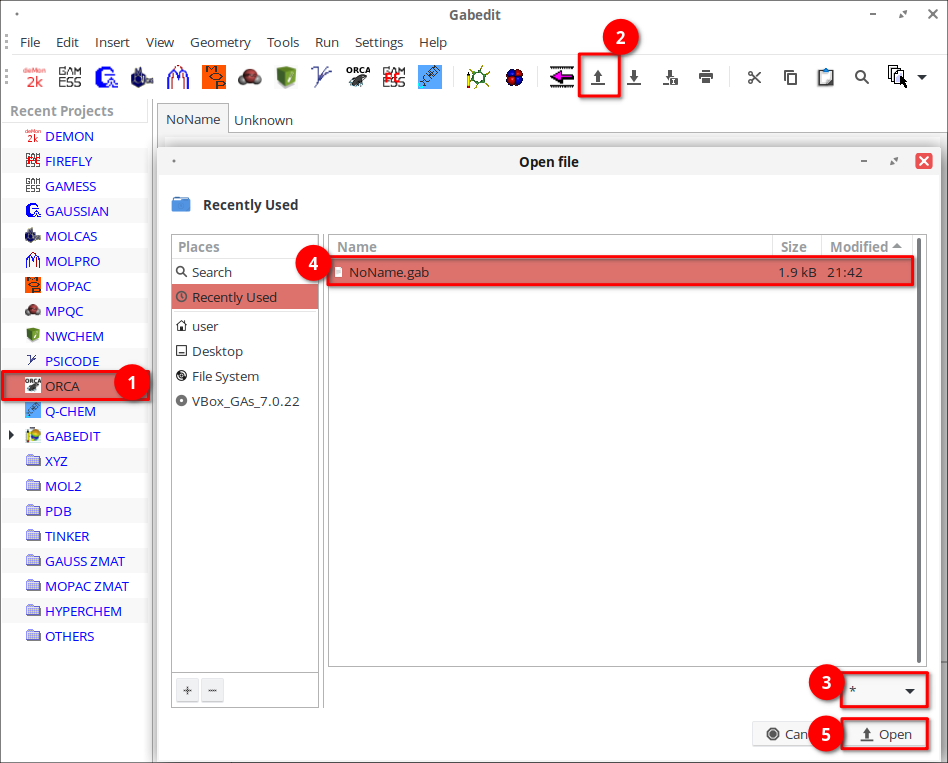
9. Дождитесь загрузки геометрии молекулы.
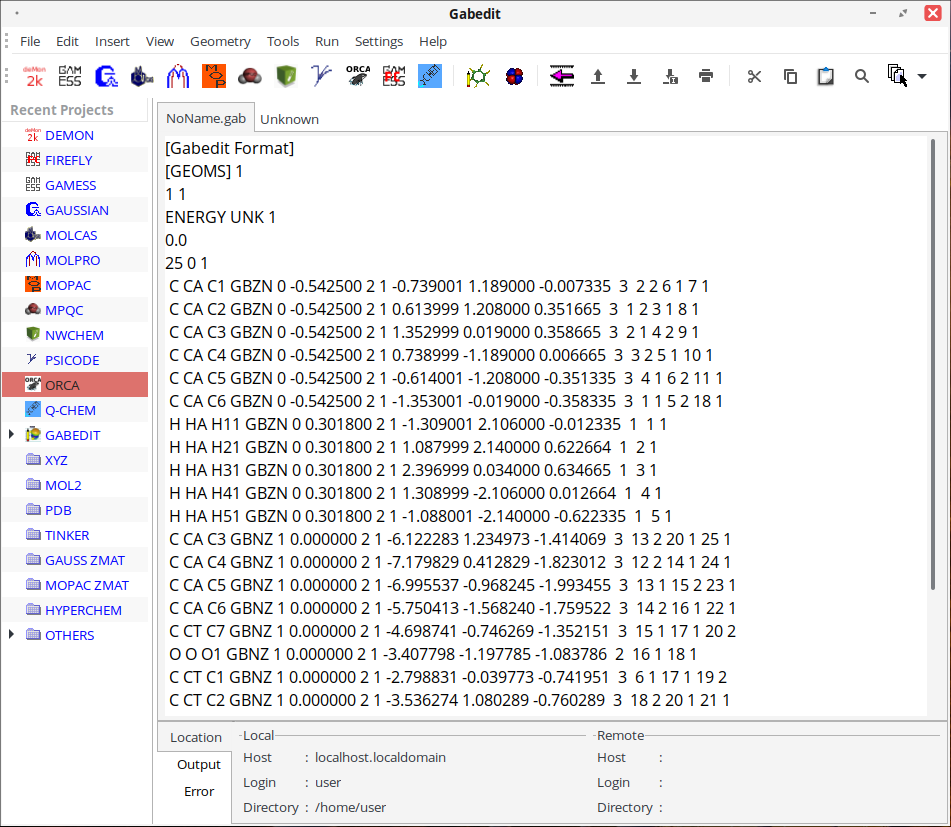
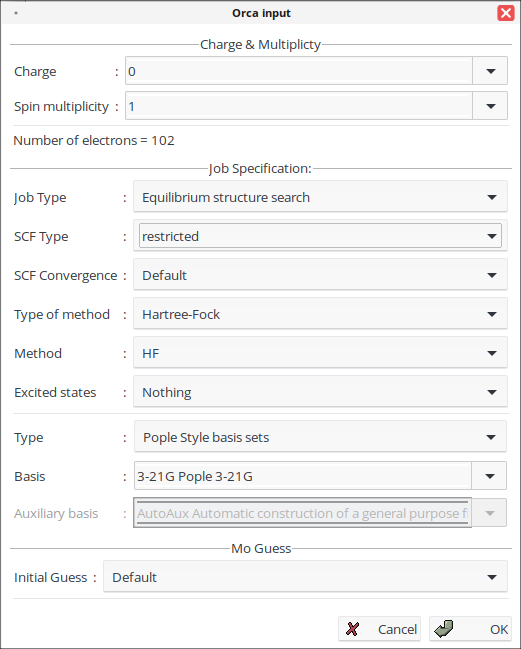
10. На панели инструментов нажмите ![]() и выберите необходимые параметры расчёта.
и выберите необходимые параметры расчёта.
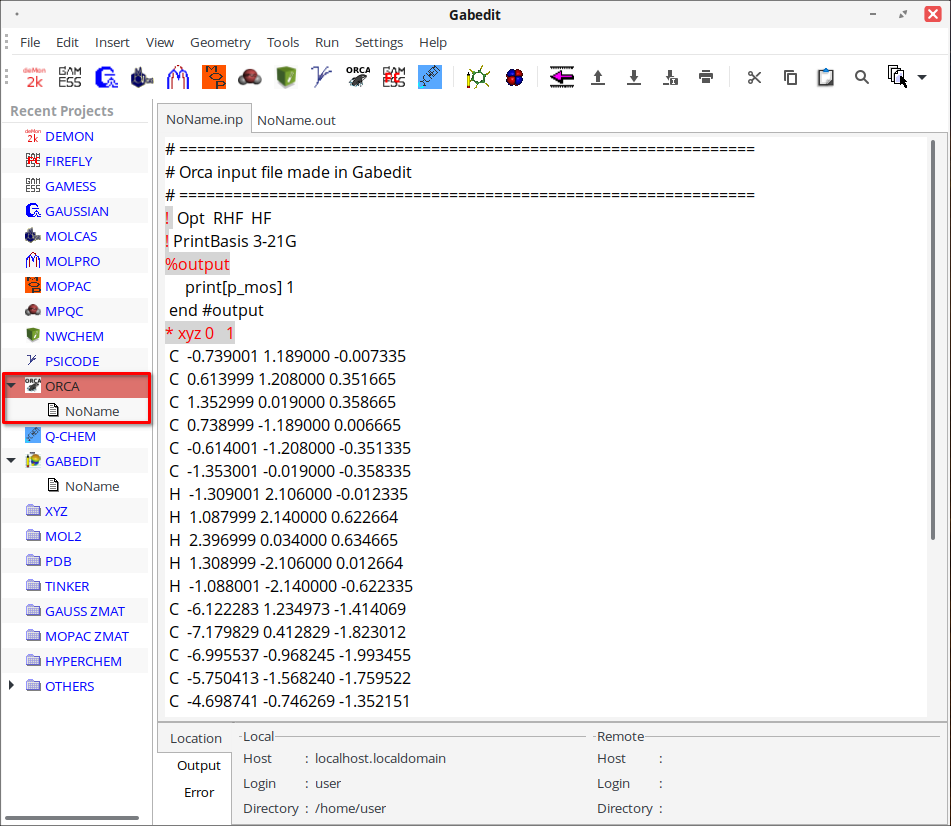
11. В меню «Run» выберите пункт «Run a Computation Chemistry program». В навигационной панели отобразится соответствующий файл расчёта.
Дата последнего изменения: 29.08.2025
Если вы нашли ошибку, пожалуйста, выделите текст и нажмите Ctrl+Enter.